Understanding the Genetic Evolution of the Pandemic H1N1 Virus
On March 28, 2009, a nine-year-old girl visited a clinic near San Diego with a fever and cough. Similar cases of febrile respiratory illness followed within the next two weeks in several places in California and Texas. At the same time, Mexican public health authorities were reporting an increasing number of cases of respiratory disease, some of them leading to serious complications, severe pneumonia, and death. In Mexico City, the number of patients with severe respiratory illness increased dramatically, with 854 cases of pneumonia and 59 reported deaths by April 24. None of the initial patients in the United States had traveled to Mexico before the onset of the respiratory illness.
The analysis of data relating to the first cases, released on April 21 by the Centers for Disease Control and Prevention, indicated that the respiratory illness was caused by an influenza virus, similar to the H1N1 viruses that were circulating in pigs in North America. Genetically and antigenically, swine H1N1 viruses are sufficiently different from human seasonal viruses, making vaccination or previous exposure to seasonal viruses unlikely to provide protection. In addition, none of those infected seemed to have been in proximity to pigs, suggesting that the emergent virus was able to transmit from human to human. The same swine origin H1N1 influenza virus was isolated in specimens from patients in Mexico. In contrast to the seasonal influenza, the new virus afflicted young and healthy adults preferentially.
At the end of April, it was clear that a novel influenza virus was spreading in North America from human-to-human transmission, causing febrile respiratory illness, and in some cases, pneumonia and death. Mexican authorities reacted energetically by canceling classes and public events. In April, the World Health Organization (WHO) issued an alert that the new strain had pandemic potential. By April 28, mild cases were reported in Canada, Spain, Israel, the United Kingdom, and New Zealand. On June 11, WHO officially declared the first pandemic of the twenty-first century.
Influenza A viruses, including seasonal and pandemic H1N1 viruses, are RNA viruses with an enormous capacity for evolving and diversifying. The size of a human genome is three billion bases, with coding for more than twenty thousand genes. We make a few copies of ourselves (our children) every twenty to thirty years, and these copies are very similar to us (the mutation rate in humans is 10-8 per replication per base). In contrast, RNA viruses, like flu or HIV, are very small (10,000 bases and only 10 genes), produce millions of copies every day, and their mutation rate is four orders of magnitude higher that humans. Humans and RNA viruses demonstrate two distinct strategies that evolution has followed: humans produce a small number of high-fidelity copies, while RNA viruses produce an enormous quantity of lousy copies. But despite their apparent simplicity, with only ten proteins, these viruses are able to hijack and use the complex machinery of the cell.
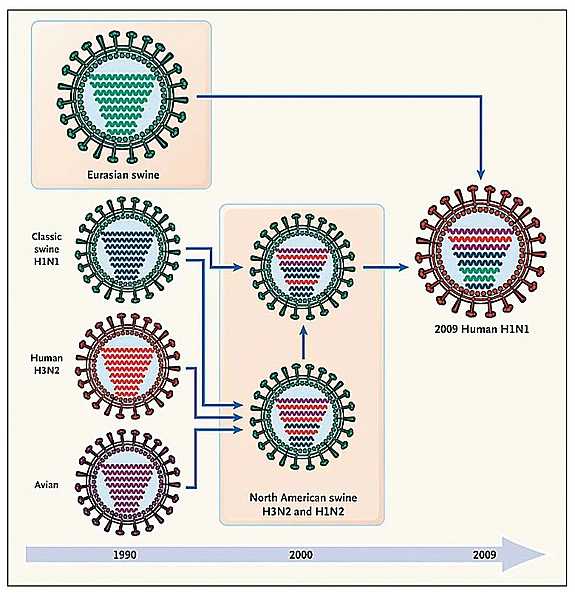
In addition to mutations, influenza viruses present another method of evolution called reassortment. The flu genome is made of eight different pieces, called segments. When two different viruses co-infect the same cell, they can produce a progeny containing the genetic material of both parental strains. This process is at the heart of pandemics, when novel viruses generated by reassortment are able to transmit from human to human.
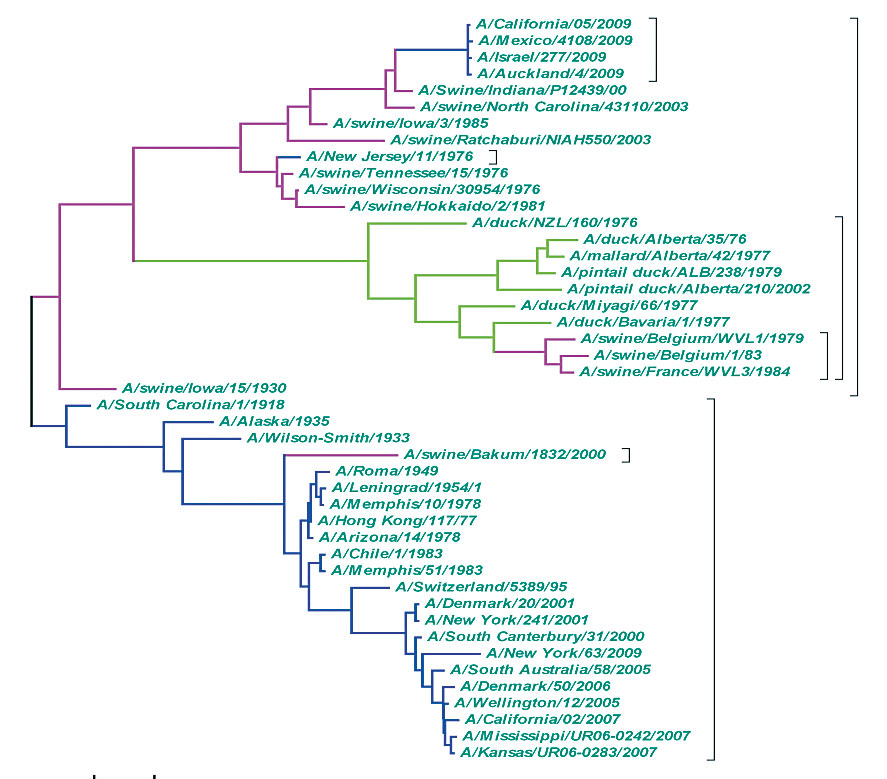
The recent H1N1 virus arose as a complicated reassortment process whose story is still not fully elucidated. By comparing the new virus to the 10,000 different genomes that have been collected and sequenced since 1918, we are able to trace back its ancestors. The new virus is related to swine viruses isolated in two continents, North America and Eurasia. The North American ancestors were isolated in pigs between 1998 and 2002 and were the result of a reassortment between swine, avian, and human viruses, the so-called triple reassortment at the end of the nineties. The origin of the Eurasian ancestors is more mysterious. They were found in Europe in the early nineties, and since then the data is sparse. How, when, and where these two ancestors met is still an open question, which probably will only be solved with a better worldwide surveillance system.
As previously mentioned, this virus is evolving and diversifying. More than a thousand viruses have been isolated and sequenced around the globe. The viruses from New York, Tokyo, Paris, Mexico, Argentina, and other locations show a very similar genome with subtle differences. By taking all this information into account, we can estimate that the most recent common ancestor to the H1N1 viruses that are currently spreading in the world is very recent, probably from the end of January or the beginning of February 2009, which is compatible to the first reported cases in Mexico and the United States. The viruses from these different locations show a strong geographic correlation, suggesting local spread in many different places around the globe, and an increasing diversity.
At the moment, the most effective strategy to fight influenza viruses is vaccination. The first step in the process of creating an influenza vaccine is to choose a virus that we think will represent the circulating virus in the near future. The high evolutionary speed, obtained through mutations and reassortments, is a very successful strategy that makes RNA viruses, like flu, very hard to predict. However, there are several lessons that we, as a species, are learning, including awareness that basic science provides the best guidance for implementing effective public health measures. We need an effective worldwide system of surveillance for emerging viruses, to map and to identify mutations that could confer resistance to drugs or vaccines, to learn how to assess the transmissibility of a pathogen in humans, and to identify the molecular factors that determine the virulence of a pathogen.